cleanUrl: "about-macs2"
description: "ChIP-seq 데이터 peak calling에 사용하는 도구인 MACS2 사용법을 정리합니다."
Mapped reads (.bam) 파일을 갖고 있다고 가정한다. (36bp짜리 짧은 read들 mapping에는 bowtie1이 좋고, 긴 read들 mappin에는 bowtie2나 bwa-mem이 좋다고 한다. 특히 bwa-mem은 error rate이 높을 때도 꽤 잘 작동한다고 한다.)
보통 ChIP-seq 실험에는 IP하지 않은, 즉 antibody 처리 안 한 control (input) sample과, 관심 있는 protein에 대한 antibody 처리를 한 IP sample 두 개를 사용한다. → 현재 control.bam과 IP.bam 두 파일이 있다고 하자.
https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html 참조
WXS에서와 같이 exact same location에 있는 read들을 하나로 친다.
→ 결국 그냥 peak calling 전에 duplicate을 없애는 게 가장 좋다. Differential binding analysis 할 때는 duplicate을 retain해도 좋고, repetitive region에 binding하는 것이 예상된다면 duplicate이라 multimapped read를 retain하는 게 좋다.
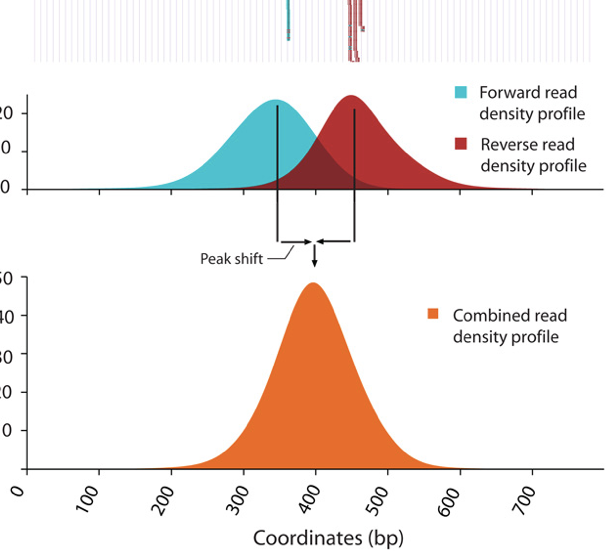
True binding site 주변의 read density를 보면 bimodal한 enrichment pattern이 나타난다.
MACS는 이러한 bimodal한 패턴을 보고 각 봉우리가 얼마나 shift되어야 '잘 합쳐진' 하나의 peak을 형성할지 모델링을 통하여 알아낸다.
가장 먼저, 어떤 지역을 분석할지를 정하는 게 필요하다. MACS는 이 과정에서 오직 ChIP 샘플만을 사용해서 (input/control 말고) significantly enriched region을 찾아낸다. bandwidth 길이의 슬라이딩 윈도우를 가지고 쭉 스캔하면서 genome에 random 하게 read가 mapping되었을 때의 density보다 mfold 배 이상 enrich되어 있는 지역을 골라내는 것이다.
그 다음, 이러한 high-quality peak 중 1000개를 random sample한다. 그 다음, positive (+) read와 negative (-) read를 분리하고, 각각을 따로 생각하여 두개의 봉우리 (mode)를 찾아낸다. 두개의 봉우리 사이의 거리를 d라고 정의하면 이것이 estimated fragment length가 된다. 각 read의 3' 방향으로 d/2 만큼 이동하면 그곳이 가장 그럴듯한 protein-DNA interaction site가 된다. → 1000개의 샘플을 가지고 d를 찾아내는 것이 핵심!
Input과 IP 샘플에서 sequencing depth가 차이가 나는 경우 MACS는 두 샘플의 total read count가 같아지도록 linear scale한다. 기본적으로 read count가 많은 샘플이 scaled down된다.
각 read를 d/2만큼 움직인 다음 MACS는 다시 2d 길이의 윈도우를 가지고 스캔하여 candidate peak를 찾아낸다. Genome 상의 read 분포는 푸아송분포로 모델링될 수 있다. 이때 lambda 는 그 window에서의 read 개수의 기댓값이다.